Vitamin D has gained enormous notoriety over the past decade, primarily due to correlation studies finding that a large portion of the population is vitamin D-deficient. Since then, study after study have found correlations between vitamin D and disease states such as heart disease and Alzheimer’s. Vitamin D is a steroid-based molecule, and its classification as a vitamin occurred in 1920, due to the notion that a lack of vitamin D can cause rickets, thus its intake is vital for health. However, more recently, it was discovered that vitamin D is multifaceted, that is, vitamin D has actions on calcium and bone homeostasis, sex hormone production, and inflammation processes. The structure of vitamin D is more similar to pregnenolone, a cholesterol derivative, than to vitamin K and other vitamins. Since vitamin D is synthesized from cholesterol, it is a fat-soluble molecule able to cross the cell membrane and bind to nuclear receptors that act to influence gene transcription.
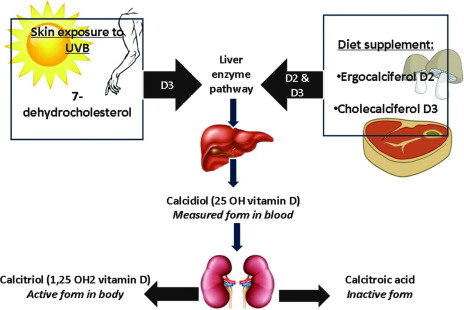
Vitamin D can be taken in from the diet; it is usually found in fish; it can be endogenously produced when the skin is exposed to ultraviolet light. After the conversion from UV light, enzymes produced and released from the liver and kidneys can convert the inactive form to the active vitamin D3 or calcitriol. In the active form, calcitriol can bind to the vitamin D receptor (VDR), which may interact with other receptors such as the retinoid X receptor (RXR), which can control the transcription of certain genes. Vitamin D plays a major role in calcium-phosphorus homeostasis at the absorption and transcriptional level, making it a multifaceted molecule.
There have been studies conducted throughout the decade that explore the actuality of previously conducted correlation studies. The model “one-size fits all” is a statement that holds true, not only in certain regions of science but in all matters of exploration. With the advancement of population and genetic studies, it is becoming increasingly known that circulating levels of vitamin D are different in different groups of people. From a logical perspective, the ancestors living in the northern regions were exposed to less UV light than ancestors living near the equator. In addition, clothing needed to keep warm oftentimes renders this UV light useless in the winter months. Also, dietary sources of vitamin D are not as abundant as previously thought, and for a hunter-gatherer group, it would be logical to think these ancestors were not receiving what we deem today a recommended daily intake of vitamin D. Besides the fact that fat-soluble vitamins can be stored in adipose tissue for long-term use, including vitamin D, genetics, and epigenetics point to another solution as to why vitamin D deficiency did not severely impact our ancestors. Genetics and epigenetics allow receptors to elicit a response intensity, such that low levels of vitamin D in a northern ancestor could facilitate an adequate response intensity, similar to that of high vitamin D in an ancestor closer to the equator. The notion that a reference range is adequate is nonsensical and has led to people taking vitamin D supplements that may contain much higher levels of vitamin D than needed, possibly harming their health.
With the necessary background, this journal review aims to investigate a recent article focusing on the negative impact of vitamin D supplementation on Alzheimer’s disease. Alzheimer’s disease (AD) is a rather complex disease, and even more so after recent scandals involving the pathology of the disease. The pathology of AD is rather extensive, but briefly, it is a neurodegenerative disease in which amyloid beta and tau tangles begin to dominate, cause inflammation, and eventually lead to neuronal death. This cell death, or infiltration of foreign proteins, causes signal disruption, which is clinically referred to as memory loss. The mechanisms behind Alzheimer’s disease are extremely complex and recent findings indicate that vitamin D levels may be decreased because of Alzheimer’s onset instead of what was previously described, which was that vitamin D deficiency causes Alzheimer’s.
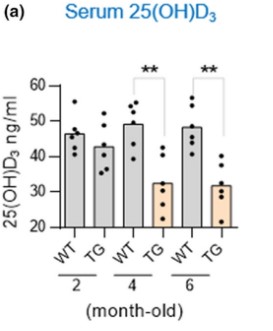
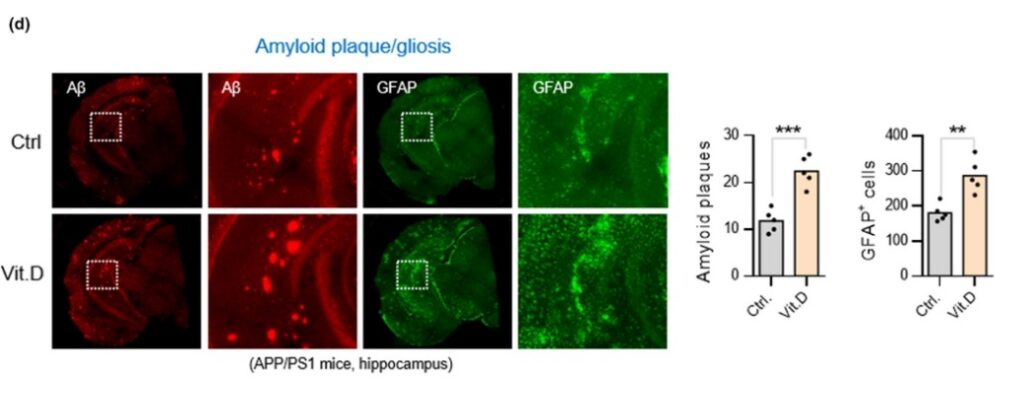
This study is very comprehensive encompassing multiple animal models and a large population study investigating VD and AD. The first model acted to investigate whether VD deficiency is caused by AD or if, as correlation studies concluded, VD deficiency causes AD. To determine this, two groups were established, a control group and an APP/PS1 Alzheimer’s group (TG). The researchers took serum vitamin D levels at 2, 4, and 6 months old to determine at which point VD. The results of this experiment showed that as AD progresses, serum VD decreases, suggesting that AD causes VD deficiency. Immunofluorescence showed that there are significantly more amyloid beta plaques in Alzheimer’s mice given a vitamin D supplement when compared to mice fed a normal diet. This trend also follows with Glial fibrillary acidic protein (GFAP), a marker for AD progression. Next, the researchers used a western blot, which is a technique used to determine if a protein is expressed. The western blot was looking at amyloid beta protein, beta secretase-1 (BACE), and a control protein GAPDH. The results indicate that in the hippocampus of Alzheimer’s-induced mice, a diet supplemented with VD resulted in higher amyloid beta plaques and BACE protein levels. To test if VD supplementation led to worse cognitive impairment in AD, a Morris water maze test was conducted and the results demonstrate that VD supplementation worsened the time of escape.
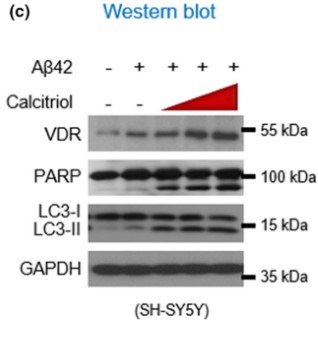
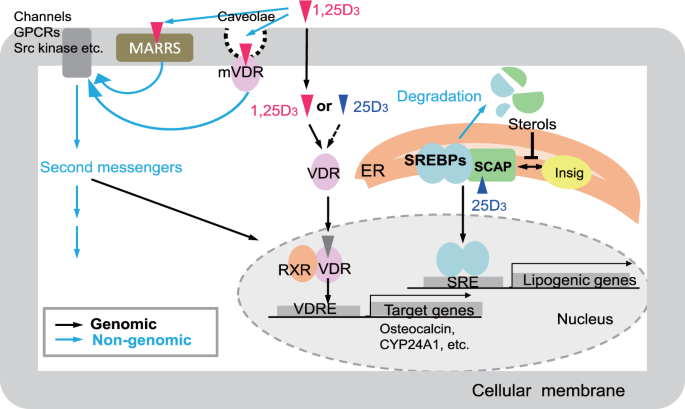
After these results, the authors then concluded that indeed the animal models pointed to the fact that AD causes VD deficiency and that supplementing with VD amidst Alzheimer’s seems to worsen the marker proteins and behavior. The next step of this study was to investigate by which mechanism is AD using or causing VD deficiency and how increasing VD via supplementation worsens AD progression. Under normal physiological conditions, when VD binds to the VDR, it binds to the retinoid X receptor, which then activates transcription via promoting transcription factor colocalization. However, existing research conducted by this group demonstrated that AD conditions may cause the VDR when bound by VD to form a complex with p53. This p53-VDR complex seems to promote pathological conditions within the cell.
To determine if the hypothesis is correct the authors decided to test whether VD supplementation improved VDR-RXR complex formation in physiological or pathophysiological conditions. The results from this experiment showed that VD supplementation in AD conditions does not rescue VDR-RXR formation. However, in normal physiological conditions, VD supplementation increased VDR-RXR complex levels two-fold compared to physiological conditions given no VD supplement. The next two western blots show that in hippocampal cells of AD mice given VD supplementation, a p53-VDR complex becomes predominant. The next few graphs demonstrated that AD complications are increased due to the formation of a VDR-p53 complex because when cells are treated with a PTF-alpha, a p53 inhibitor, amyloid beta plaques and GFAP-positive cells are totally alleviated when compared to cells not treated with the inhibitor.
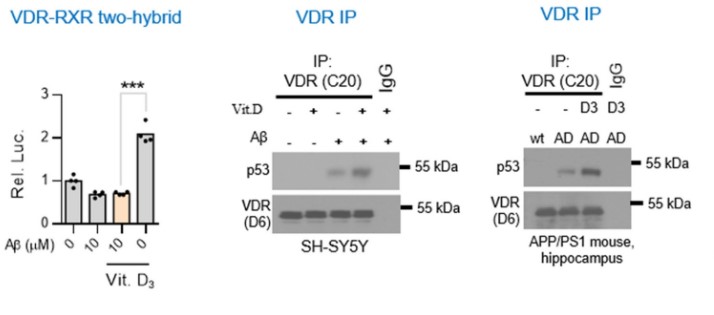
So far, the paper has concluded some major points about AD that have been quite controversial. Firstly, the data from this paper hints at the fact that VD supplementation increases GFAP-positive cells and amyloid beta plaques when supplementation is started during the onset of AD. Secondly, it also seems like VD supplementation during the onset of AD increases the formation of VDR-p53 complexes which play a role in AD pathophysiology.
To elevate the claims the authors made, a large population human study using 14,648 subjects over the age of 65 and dementia-free. The findings from the large-population study demonstrate that a low dose of VD did not impact the incidence of dementia. However, a large dose or medium dose of VD significantly elevated the risk of dementia. A similar study using 980 patients with preexisting dementia were followed for 11 years and compared the effects of a VD supplementation on mortality. It was found that a high dose of VD increased the mortality of dementia by 2-fold. However, it was found that a medium or low dose did not impact mortality. The findings of the large-population study need further validation, better controls, and a clinical trial to be conclusive. The findings could be indicative of the issues with a VD supplement in patients with preexisting dementia or patients prone to dementia. In conclusion, a vitamin D supplement in the elderly seems to have beneficial impacts on bone health but may negatively affect neurological health. The in vitro and in vivo experiments in this study are promising and require more validation because several other papers have found conflicting results yet are not as comprehensive as this research.

Hello everyone,
My name is Joshua Giblin. I am a post-bachelor researcher/research technician at USC. My interests range from nutrition to nanomedicine and also practical science to improve everyday life. Through this blog, I aim to communicate practical scientific research and present it to curious individuals so that an educated decision can be made. Thank you for reading the blog and showing your support.
Literature cited
- Chatterjee, P., Pedrini, S., Stoops, E., Goozee, K., Villemagne, V. L., Asih, P. R., Verberk, I. M. W., Dave, P., Taddei, K., Sohrabi, H. R., Zetterberg, H., Blennow, K., Teunissen, C. E., Vanderstichele, H. M., & Martins, R. N. (2021). Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Translational Psychiatry, 11(1), Article 1. https://doi.org/10.1038/s41398-020-01137-1
- Lai, R.-H., Hsu, C.-C., Yu, B.-H., Lo, Y.-R., Hsu, Y.-Y., Chen, M.-H., & Juang, J.-L. (2022). Vitamin D supplementation worsens Alzheimer’s progression: Animal model and human cohort studies. Aging Cell, 21(8), e13670. https://doi.org/10.1111/acel.13670
- Nishikawa, M., Yasuda, K., Takamatsu, M., Abe, K., Okamoto, K., Horibe, K., Mano, H., Nakagawa, K., Tsugawa, N., Hirota, Y., Horie, T., Hinoi, E., Okano, T., Ikushiro, S., & Sakaki, T. (2020). Generation of novel genetically modified rats to reveal the molecular mechanisms of vitamin D actions. Scientific Reports, 10(1), Article 1. https://doi.org/10.1038/s41598-020-62048-1